Preservation of developmental spontaneous activity enables early auditory system maturation in deaf mice
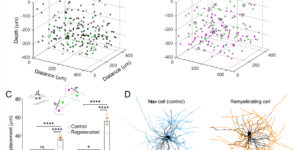
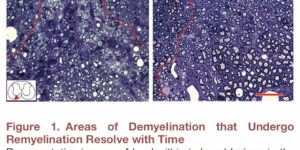
Generation of oligodendrocytes in the adult brain enables both adaptive changes in neural circuits and regeneration of myelin sheaths destroyed by injury, disease, and normal aging. This transformation of oligodendrocyte precursor cells (OPCs) into myelinating oligodendrocytes requires processing of distinct mRNAs at different stages of cell maturation. Although mislocalization and aggregation of the RNA-binding protein, TDP-43, occur in both neurons and glia in neurodegenerative diseases, the consequences of TDP-43 loss within different stages of the oligodendrocyte lineage are not well understood. By performing stage-specific genetic inactivation of Tardbp in vivo, we show that oligodendrocyte lineage cells are differentially sensitive to loss of TDP-43. While OPCs depend on TDP-43 for survival, with conditional deletion resulting in cascading cell loss followed by rapid regeneration to restore their density, oligodendrocytes become less sensitive to TDP-43 depletion as they mature. Deletion of TDP-43 early in the maturation process led to eventual oligodendrocyte degeneration, seizures, and premature lethality, while oligodendrocytes that experienced late deletion survived and mice exhibited a normal lifespan. At both stages, TDP-43-deficient oligodendrocytes formed fewer and thinner myelin sheaths and extended new processes that inappropriately wrapped neuronal somata and blood vessels. Transcriptional analysis revealed that in the absence of TDP-43, key proteins involved in oligodendrocyte maturation and myelination were misspliced, leading to aberrant incorporation of cryptic exons. Inducible deletion of TDP-43 from oligodendrocytes in the adult central nervous system (CNS) induced the same progressive morphological changes and mice acquired profound hindlimb weakness, suggesting that loss of TDP-43 function in oligodendrocytes may contribute to neuronal dysfunction in neurodegenerative disease.