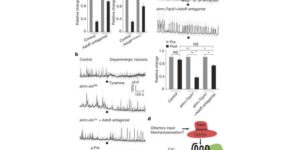
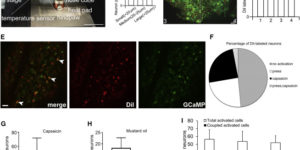
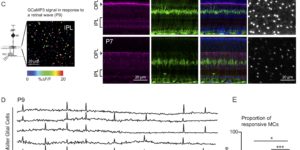
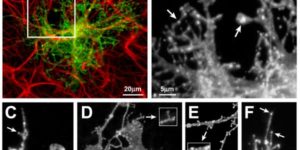
Activity-dependent switch of GABAergic inhibition into glutamatergic excitation in astrocyte-neuron networks.
Interneurons are critical for proper neural network function and can activate Ca2+ signaling in astrocytes. However, the impact of the interneuron-astrocyte signaling into neuronal network operation remains unknown. Using the simplest hippocampal Astrocyte-Neuron network, i.e., GABAergic interneuron, pyramidal neuron, single CA3-CA1 glutamatergic synapse, and astrocytes, we found that interneuron-astrocyte signaling dynamically affected excitatory neurotransmission in an activity- and time-dependent manner, and determined the sign (inhibition vs potentiation) of the GABA-mediated effects. While synaptic inhibition was mediated by GABAA receptors, potentiation involved astrocyte GABAB receptors, astrocytic glutamate release, and presynaptic metabotropic glutamate receptors. Using conditional astrocyte-specific GABAB receptor (Gabbr1) knockout mice, we confirmed the glial source of the interneuron-induced potentiation, and demonstrated the involvement of astrocytes in hippocampal theta and gamma oscillations in vivo. Therefore, astrocytes decode interneuron activity and transform inhibitory into excitatory signals, contributing to the emergence of novel network properties resulting from the interneuron-astrocyte interplay.