Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina.
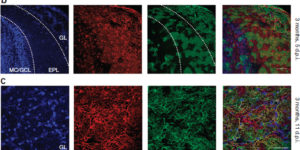
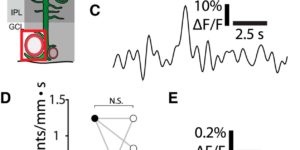
The brain is critically dependent on the regulation of blood flow to nourish active neurons. One widely held hypothesis of blood flow regulation holds that active neurons stimulate Ca(2+) increases in glial cells, triggering glial release of vasodilating agents. This hypothesis has been challenged, as arteriole dilation can occur in the absence of glial Ca(2+) signaling. We address this controversy by imaging glial Ca(2+) signaling and vessel dilation in the mouse retina. We find that sensory stimulation results in Ca(2+) increases in the glial endfeet contacting capillaries, but not arterioles, and that capillary dilations often follow spontaneous Ca(2+) signaling. In IP3R2(-/-) mice, where glial Ca(2+) signaling is reduced, light-evoked capillary, but not arteriole, dilation is abolished. The results show that, independent of arterioles, capillaries actively dilate and regulate blood flow. Furthermore, the results demonstrate that glial Ca(2+) signaling regulates capillary but not arteriole blood flow.