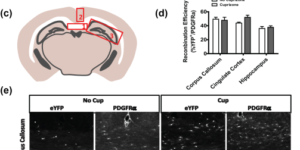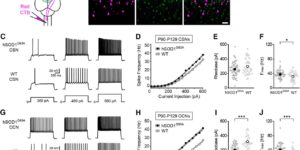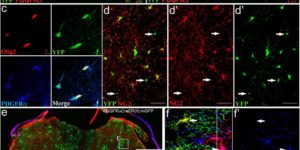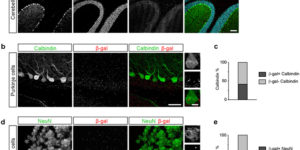
Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex.
Oligodendrocyte generation in the adult CNS provides a means to adapt the properties of circuits to changes in life experience. However, little is known about the dynamics of oligodendrocytes and the extent of myelin remodeling in the mature brain. Using longitudinal in vivo two-photon imaging of oligodendrocytes and their progenitors in the mouse cerebral cortex, we show that myelination is an inefficient and extended process, with half of the final complement of oligodendrocytes generated after 4 months of age. Oligodendrocytes that successfully integrated formed new sheaths on unmyelinated and sparsely myelinated axons, and they were extremely stable, gradually changing the pattern of myelination. Sensory enrichment robustly increased oligodendrocyte integration, but did not change the length of existing sheaths. This experience-dependent enhancement of myelination in the mature cortex may accelerate information transfer in these circuits and strengthen the ability of axons to sustain activity by providing additional metabolic support.